Drug Discovery
Exploring the kinome to unlock new possibilities
With our sights set on discovery, Aclaris is currently exploring the kinome, a subset of the human genome that consists of a collection of approximately 518 protein kinases, one of the largest of all human gene families, responsible for signal transduction controlling cellular responses.
Classified into eight major groups based on their structural similarity to each other, kinases are key regulators of cell function for many cell processes. By transferring phosphates to other molecules, kinases can induce a cellular response to environmental cues. Dysregulation and/or activating/blocking mutations in kinases can disrupt normal cell signaling and lead to diseases ranging from autoimmune diseases to diabetes and cancer, making them important targets for drug development.
Currently, there are a number of approved kinase inhibitors on the market, such as Xeljanz®* (tofacitinib), Gleevec®* (imatinib mesylate), and Tarceva®* (erlotinib). However, these drugs only target a small fraction of the kinome, with many clinically relevant kinases lacking validated inhibitors.
We’re focused on the design and development of kinase inhibitors that target key enzymes involved in chronic inflammation, autoimmune disease, or the regulation of cancer growth, survival and metastasis.
*All trademarks are the property of their respective owners.
KINect® platform: proprietary discoveries through kinome innovation
Our proprietary KINect® platform accelerates the identification of drug candidates through a unique combination of our proprietary chemical library of kinase inhibitors, our expertise in structure-based drug design, and our custom kinase assays.
Central to the KINect® platform is our novel chemical library of several hundred compounds specifically designed to target non-catalytic cysteine residues near the adenosine triphosphate (ATP) binding site of more than 300 kinases.
Using powerful drug modeling software, we are able to elaborate the structure of viable drug-like compounds culled from our library to optimize reversible binding to the target kinase and allow them to selectively form a covalent bond with the cysteine residue near the ATP site on the specific kinase target.
We then assess the function of the newly created drug candidates with physiologically relevant custom assays that effectively translate to human diseases.
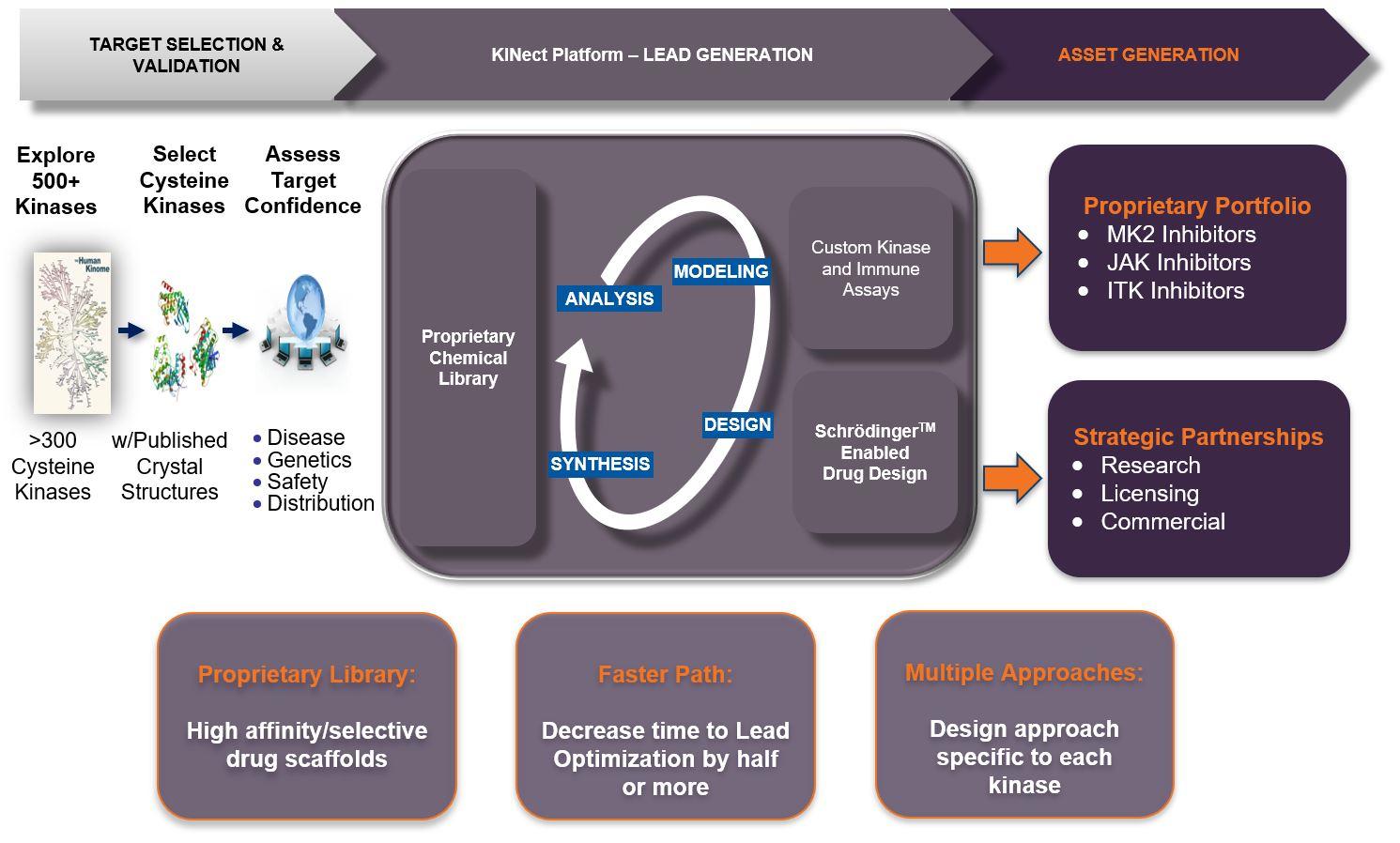
The KINect® platform allows us to discover novel drug candidates in a fraction of the time of traditional approaches: 1-2 months to identify and optimize novel lead chemical series that bind to a target of interest versus more than six months to years for traditional approaches that lack a tailored library.
The platform will generate inhibitors with fit-for purpose mechanisms ranging from reversible, to reversible-covalent to irreversible-covalent. These drug candidates have the potential to overcome kinome selectivity and biochemical inefficiency issues faced by other approaches for validated kinase targets.
Clinical Development Compounds
ATI-1777: Investigational Soft Topical JAK1/JAK3 Inhibitor
Potential therapeutic targets: atopic dermatitis; potentially other dermatologic conditions.
Soft JAK inhibitors (ATI-1777) are designed to be topically applied and active in the skin, but may be rapidly metabolized and inactivated when they enter the bloodstream, which may result in significantly reduced systemic exposure. These tissue-restricted properties may result in efficacy with potential minimization of systemic safety issues. The JAK family of kinases are a subgroup of non-receptor tyrosine kinases that are essential in transducing signals originating from cytokine receptors. JAK inhibitors are approved to treat inflammatory and autoimmune disorders.
ATI-2138: Investigational Oral Covalent ITK/JAK3 Inhibitor
Potential therapeutic targets: T cell-mediated autoimmune diseases.
ATI-2138 is an investigational oral covalent ITK/JAK3 inhibitor. ITK and JAK3 are important targets in immune regulation.
ITK inhibitors target the non-receptor tyrosine kinase, ITK, within the T-cell receptor (TCR) signaling pathway, thereby interfering with the development and effector function of immune system T cells. ITK is a key signaling kinase downstream of the TCR and as such, regulates IL17 and IFNγ expression. The potential combination effect of inhibiting T cell receptors (inhibiting T cell maturation and activation) and IL17 production means an ITK inhibitor may function as a “small molecule anti-IL17”, but with broader immunomodulatory activity.
JAK3 regulates key cytokines that signal through the common gamma chain receptor such as IL2, IL4, IL7, IL9, IL15 and IL21.
A dual ITK/JAK3 inhibitor could result in additive or synergistic anti-inflammatory activity that may be beneficial in a number of autoimmune and inflammatory diseases.
Zunsemetinib (ATI-450): Investigational Oral MK2 Pathway Inhibitor
Potential therapeutic targets: metastatic breast cancer (MBC); pancreatic ductal adenocarcinoma (PDAC).
MBC: Phosphorylated MK2 is upregulated in primary tumors and metastatic bone lesions from MBC patients. MK2 is responsible for the production of a subset of critical pro-tumorigenic factors secreted by the stromal microenvironment to support tumor growth and metastasis. Additionally, MK2 drives both metastatic and chemotherapy induced bone loss in MBC patients through, at least in part, its role in RANKL biology and osteoclast production and activation. In preclinical studies, zunsemetinib has been demonstrated to impact murine models of MBC through inhibition of tumor growth and metastasis along with bone preservation.
PDAC: Phosphorylated MK2 is highly expressed in PDAC tissue and expression levels are directly associated with poor outcomes in patients with PDAC. The current first and second line standard of care for PDAC patients is FOLFIRINOX combination chemotherapy. Irinotecan and its metabolite, SN-38, are the main drivers of cancer cell apoptosis associated with FOLFIRINOX. The effectiveness of FOLFIRINOX is limited by pro-survival resistance mechanisms that are driven through SN-38 activation of the MK2 pathway and phosphorylation of two direct MK2 substrates, HSP-27 and Beclin-1. In both patient derived xenografts and in the autochthonous genetic KPPC model of PDAC in mice, zunsemetinib has demonstrated that it blocks phosphorylation and activation of HSP-27, induces tumor cell killing and enhances the efficacy of FIRINOX (a version of FOLFIRINOX used in murine models).